
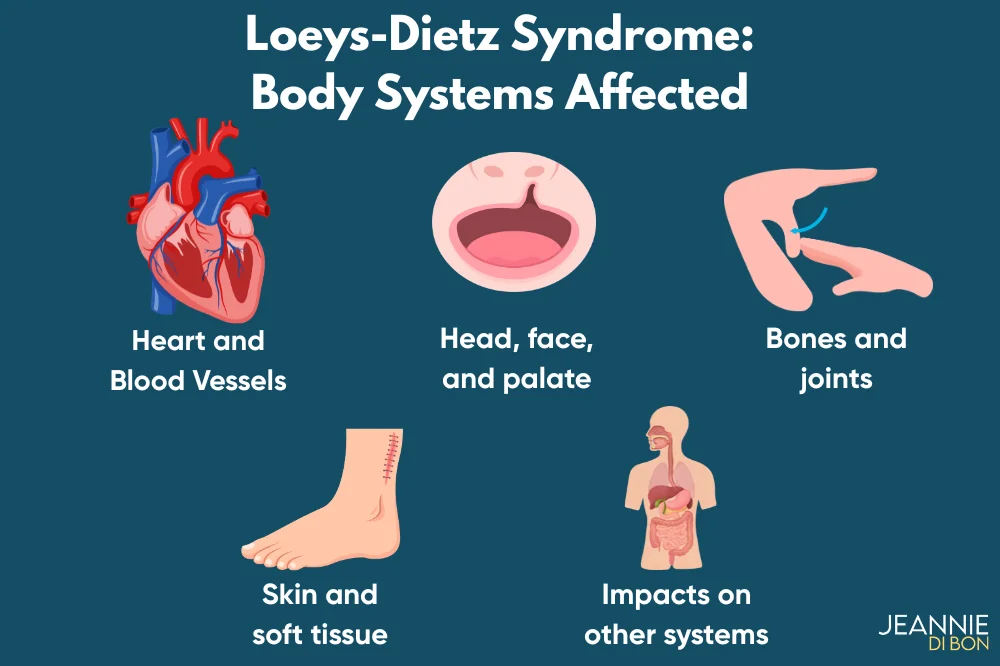
Estimated reading time: 7 minutesLoeys-Dietz syndrome (LDS) is a rare inherited connective tissue disorder that can affect the heart and blood vessels, bones and joints, face and palate, skin, and several other body systems.
When it comes to navigating the realities of living with a rare connective tissue disorder, Maggie Buckley is an expert in many ways. She is a patient herself and a long-time patient advocate, educator, and member of the EDS and Loeys-Dietz communities.
I asked Maggie to share that knowledge, combining decades of advocacy work with her own lived experience of these conditions. I’m delighted to share this article by Maggie on LDS to help raise awareness of this condition and patients’ lived experiences.
Key Takeaways
- Loeys-Dietz syndrome is a rare inherited connective tissue disorder caused by variants in genes that control the TGFβ signaling pathway, affecting blood vessels, bones, skin, and other systems.
- The biggest medical concern in LDS is the risk of aortic aneurysm and arterial dissection, which is why early diagnosis and regular imaging are important even when someone feels well.
- LDS shares features with Marfan syndrome and EDS but is a distinct condition with different genetic causes and clinical priorities.
- Diagnosis requires genetic testing alongside imaging and clinical evaluation — outward features alone are not enough to confirm or rule it out.
- Movement and exercise are possible for most people with LDS, but activity guidance should always be individualised based on vascular findings and recent imaging.
What Is Loeys-Dietz Syndrome?
Loeys-Dietz syndrome (LDS) is a rare inherited connective tissue disorder caused by variants in genes that control the TGFβ signaling pathway. This system is used by the body to help guide growth, repair, and tissue maintenance.
When this pathway is disrupted, connective tissue can become more fragile or develop differently, which can affect blood vessels, the skeleton, the skin, the face and palate, and sometimes the immune, digestive, and nervous systems.
The biggest medical concern is often the risk of aortic aneurysm, arterial tortuosity (elongation, twisting, or kinking of arteries), and arterial dissection (a tear along the inside lining of the artery), which is why early diagnosis and regular imaging are important.
Because the condition can look very different from one person to another, care is usually most effective when it is individualized and coordinated across specialties rather than managed by only one clinician.
Quick facts about Loeys-Dietz syndrome
- LDS was first recognized as a distinct condition in 2005 by Dr. Bart Loeys and Dr. Harry Dietz
- In plain language, TGFβ signaling is part of the body’s communication system for building and maintaining tissues; when one of the related genes is altered, blood vessels and other connective tissues may not develop or heal normally.
- LDS can affect the aorta and other arteries, bones and joints, the face and palate, skin, eyes, ears, lungs, digestive tract, and sometimes immune-related health.
- Older materials often refer to five subtypes, but current classification recognizes six genes involved in TGFβ signaling associated with LDS, and the exact pattern can differ depending on the gene involved.
What Are the Symptoms of Loeys-Dietz Syndrome?
The symptoms of Loeys-Dietz syndrome are wide-ranging and can vary a lot between individuals, even within the same family. Some people are identified because of blood vessel findings on imaging, while others first notice skeletal, skin, craniofacial, digestive, allergic, or joint-related features.
The most commonly reported symptoms by body system include:
- Heart and blood vessels: aortic root enlargement, aneurysms in other arteries, arterial tortuosity, and risk of dissection or tear.
- Head, face, and palate: widely spaced eyes, a bifid or broad uvula, cleft palate, and sometimes craniosynostosis or other craniofacial differences.
- Bones and joints: scoliosis, chest wall differences, long fingers, joint hypermobility or instability, contractures, cervical spine issues, and foot differences such as clubfoot or flat feet.
- Skin and soft tissue: easy bruising, soft or translucent skin, wider scars, stretch marks, and hernias.
- Other possible symptoms: digestive problems, allergies or immune-related issues, hearing or vision concerns, pain, fatigue, and overlapping features with other connective tissue disorders.
How Is Loeys-Dietz Syndrome Different from Marfan Syndrome?
Loeys-Dietz syndrome and Marfan syndrome are both inherited connective tissue disorders that can cause aortic enlargement, skeletal differences, and overlapping physical features, so they can look similar at first.
The key differences are that LDS is caused by variants in TGFβ pathway genes rather than the FBN1 gene.
LDS often involves aneurysms and twisting of arteries beyond the aortic root, and may present with features such as widely spaced eyes, a bifid or broad uvula, or cleft palate.
Another important distinction is that lens dislocation is a hallmark of Marfan syndrome but is not a typical feature of LDS, and vascular risk in LDS can become serious at younger ages or at smaller aortic diameters.
How Is Loeys-Dietz Syndrome Related to Ehlers-Danlos Syndrome and HSD?
Loeys-Dietz syndrome, Ehlers-Danlos syndrome, and hypermobility spectrum disorder share some overlapping features — including joint laxity, pain, easy bruising, and soft skin — but they are distinct conditions with different underlying causes and clinical priorities.
The main difference is that LDS is especially associated with aortic aneurysm, arterial tortuosity, and characteristic craniofacial findings, while EDS is a broader group of disorders that more often centers on skin fragility, joint hypermobility, and tissue fragility, depending on the subtype.
HSD describes symptomatic hypermobility without meeting criteria for a specific heritable connective tissue syndrome.
It is possible for someone to have features that resemble more than one connective tissue disorder, and in uncommon cases, a person may have more than one diagnosis.
Because the overlap can be confusing, the best way to sort this out is through evaluation by genetics and other specialists, along with imaging and molecular testing when appropriate.
How Is Loeys-Dietz Syndrome Diagnosed?
Loeys-Dietz syndrome is diagnosed by combining personal and family history, physical findings, vascular imaging, and genetic testing.
Today, diagnosis generally depends on finding a pathogenic or likely pathogenic variant in a gene currently recognized for LDS, especially in someone with unexplained aortic root enlargement, arterial aneurysm, or tortuosity at a young age, suggestive craniofacial or skeletal features, or a family history of aneurysm, dissection, or a known LDS-related variant.
Because some people have only subtle outward features, a normal early scan does not completely rule out future risk, and relatives may also need targeted testing and follow-up.

How Is Loeys-Dietz Syndrome Treated and Monitored?
Treatment for Loeys-Dietz syndrome focuses on lowering the risk of life-threatening vascular events while also managing the other body systems the condition can affect.
There is no cure, but regular imaging, medication to reduce stress on the aorta and arteries, and preventive surgery when indicated can significantly lower the risk of serious complications.
Long-term care is usually individualized and may include regular heart imaging, scans of the arteries from head through pelvis, medicines to reduce stress on blood vessels, activity guidance, and preventive surgery when vessel size, growth rate, family history, or genetic subtype suggests higher risk.
The main components of treatment and monitoring include:
- Regular imaging: Many people need echocardiograms and periodic MRI or CT imaging to track the aorta and other arteries over time, even when they feel well.
- Medicines: Doctors often use medicines such as beta blockers or angiotensin receptor blockers to help lower stress on the aorta and arteries.
- Preventive surgery: Surgery may be recommended before an emergency happens if imaging shows concerning enlargement, fast growth, or other high-risk features.
- Care beyond the arteries: Follow-up may also include genetics, orthopedics, craniofacial care, allergy or gastroenterology care, hearing and vision support, and pregnancy planning when relevant.
- Lifelong follow-up: Because LDS can change over time, treatment and surveillance plans usually need regular review and adjustment with an experienced care team.
What Should You Ask Your Care Team About Movement and Exercise?
Movement and exercise are possible and encouraged for most people with Loeys-Dietz syndrome, but the right approach depends on individual vascular findings, medication, symptoms, and recent imaging results. The most important first step is asking your care team for guidance specific to your situation.
Ask your care team what kinds of movement are safe for your specific vascular findings, medications, symptoms, and recent imaging results.
In general, many people with LDS can stay active with individualized guidance, but exercise planning should focus on steady, controlled activity and avoid situations that sharply increase blood pressure or involve straining, heavy contact, or high injury risk.
It is also reasonable to ask how often your activity plan should be updated, whether physical therapy would help, and what warning symptoms mean you should stop and get medical advice.

Maggie’s Story
My health story began in childhood, when easy bruising, joint pain, fatigue, sprains, allergies, stomach issues, and repeated dislocations made it clear my body was not functioning typically, even before anyone could explain why.
At thirteen, an orthopedist recognized that I had a hypermobile connective tissue disorder and gave me a lasting framework for living: learn my body’s limits, stay curious, prepare carefully, and stop when my body said enough.
Over the years, I moved through physical therapy, counseling, adaptation, and a long diagnostic journey that included labels such as Ehlers-Danlos syndrome, Marfan syndrome, and Loeys-Dietz syndrome, along with growing complications including fainting, spinal problems, osteoporosis, and stroke.
Eventually, advancing genetic testing and specialist evaluation clarified that I have an ultra-rare combination of Loeys-Dietz syndrome type 5 and arthrochalasia Ehlers-Danlos syndrome, even if I do not present in a perfectly textbook way.
What has sustained me is a disciplined practice of listening to my body through movement, mindfulness, rest, and continual adjustment, without allowing the complexity of my health to define the whole of who I am.
Frequently Asked Questions About Loeys-Dietz Syndrome
Is Loeys-Dietz syndrome hereditary?
Yes. LDS is usually inherited in an autosomal dominant pattern, which means a parent with the condition can pass it to a child, but some people are the first in their family to have it because of a new genetic change.
How common is Loeys-Dietz syndrome?
LDS is considered rare. The exact frequency is not firmly established, in part because diagnosis has broadened over time as genetic testing has improved.
Can you have Loeys-Dietz syndrome without a family history?
Yes. A person can have LDS because of a de novo, or new, pathogenic variant even if no one else in the family is known to be affected.
What is a bifid uvula and why does it matter in LDS?
A bifid uvula is a uvula that looks split or broader than usual. It does not confirm LDS by itself, but it is one of the clues that can raise suspicion for the condition when it appears with vascular or other connective tissue findings.
Can children be diagnosed with Loeys-Dietz syndrome?
Yes. LDS can be diagnosed in childhood, especially when there are suggestive physical features, family history, or abnormal imaging, and early diagnosis can help guide safer monitoring and care.
Can you have both LDS and EDS?
Sometimes. Some people have overlapping symptoms, and a small number may ultimately receive more than one diagnosis, so genetics evaluation can be important when the picture is unclear.

Ms. Buckley has been a Health/Patient Advocate for over three decades while living with a chronic pain condition. She works with individuals and their family/caregivers to access appropriate healthcare services to address their most pressing needs. Maintaining a robust network with other Health Advocates enables her to refer clients to specialist Advocates as needed. She currently volunteers in many roles for the Ehlers Danlos Society, the Loeys-Dietz Syndrome Foundation Canada and the Loeys-Dietz Foundation (a division of the Marfan Foundation USA). Additionally, she has represented the voices of lived experience on several projects researching connective tissue disorders, pain management and other health topics.

No Comments